| OMIM: | 175100 |
| Diagnostik: | Sequenzierung und CNV: APC, BMPR1A, GREM1, MLH1, MSH2, MSH3, MSH6, MUTYH, NTHL1, PMS2, POLD1, POLE, PTEN, RNF43, SMAD4, STK11 |
| Material: | 2 ml EDTA-Blut (2 Röhrchen) |
| Analysezeit: | 6-8 Wochen |
| Formulare: |
Die familiäre adenomatöse Polyposis Coli (FAP) ist gekennzeichnet durch eine Vielzahl von Polypen (Adenome) des Kolon (Dickdarmes) und Rektum (Enddarmes), die sich i.d.R. vor der Pubertät zu entwickeln beginnen. Bei der klassischen FAP finden sich hundert bis mehrere tausend Polypen, bei der abgeschwächten (attenuierten) FAP liegen weniger als hundert Polypen im Dickdarm vor. Polypen sind Ausstülpungen der Darmschleimhaut. Sie sind gutartig, können sich jedoch zum Karzinom entwickeln.
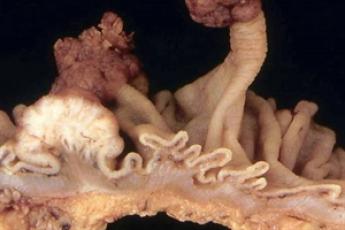
Kolon-Adenome und Karzinom
(Quelle: mit freundlicher Genehmigung von Prof. Dr. med. S. Schröder)
Die FAP gilt als Präkanzerose. Die ersten Polypen treten in der Regel mit 10-12 Jahren im Rektum oder Sigmoid auf. Ohne ärztliche Intervention entwickelt sich bei fast 100% der Patienten ein Dickdarmkarzinom, meist mit Mitte bis Ende 30.
Ca. 1% aller kolorektalen Karzinome sind auf FAP zurückzuführen. Auch Veränderungen an anderen Organsystemen können bei der FAP auftreten. Netzhauthypertrophien (Hypertrophie des retinalen Pigmentepithels) und Desmoidbildung im Bereich von OP-Narben bzw. der Papilla vateri finden sich bei einem Großteil der FAP-Patienten, aber auch Hautneoplasien, Drüsenkörperzysten des Magens oder Zahn-und Kieferanomalien sind beschrieben. Sporadisch entwickeln sich Schilddrüsenkarzinome, Dünndarmtumoren, Hirntumoren (Medulloblastome), Hepatoblastome (im Kindesalter), Magenkarzinome, Pankreaskarzinome oder auch Astrozytome. Auch Lipome, Osteome und Epidermoidzysten kommen bei den Patienten vor.
Etwa 80 Prozent der Patienten mit der klassischen Form der FAP weisen Mutationen im APC-Gen als Ursache der Erkrankung auf. Die FAP mit Mutationen im APC-Gen wird autosomal dominant vererbt, d.h. es besteht für Kinder eines Betroffenen ein Risiko von 50 Prozent, die Mutation zu erben und dann sicher im Laufe des Lebens die FAP zu entwickeln. Die Neumutationsrate beträgt etwa 25%, so dass häufig auch bei sporadischen FAP-Patienten ohne familiäre Belastung Mutationen im APC-Gen gefunden werden. Das APC-Gen ist auf Chromosom 5 lokalisiert und die Penetranz beträgt nahezu 100% (Gentest bei Darmkrebs).
Neben der klassischen FAP gibt es eine abgeschwächte Form der FAP (attenuierte FAP, AFAP), bei der sich weniger als 100 Polypen entwickeln. Häufig handelt es sich um 30-40 Polypen vor allem im rechten Dickdarm („rectal sparing“). Meist zeigt sich ein Mischbild aus hyperplastischen Polypen, Adenomen und sessilen serratierten Adenomen. Das Risiko eines Dickdarmkarzinoms liegt bei dieser Form der FAP bei ca. 80% bis zum 70.Lebenjahr. Meist tritt die Darmkrebserkrankung mit Mitte 50 auf. Bei der attenuierten FAP findet sich bei <30% der Patienten eine Mutation im APC-Gen.
Etwa zehn Prozent der Patienten mit klassischer FAP und etwa 30 Prozent der Patienten mit abgeschwächter Form weisen Mutationen im MUTYH Gen auf (MAP = MUTYH-assoziierte Polyposis). Die FAP mit Mutationen im MUTYH Gen wird autosomal rezessiv vererbt, hier besteht ein erhöhtes Wiederholungsrisiko insbesondere für Geschwister eines Betroffenen. In einigen Fällen lassen sich auch Mutationen in den Genen MSH3, NTHL1, POLD1, POLE nachweisen.
Bei nachgewiesenen Mutationen in einem der genannten Gene bzw. bei Personen mit einem hohen Risiko für eine FAP wird ein besonderes Vorsorgeprogramm bzw. z. T. die operative Entfernung des Dickdarms empfohlen.
Indikation:
- Nachweis einer erhöhten Anzahl von Polypen des Kolon und Rektum
- unklare chronische gastrointestinale Blutungen und Diarrhöen
Bei nachgewiesener Mutation im APC-Gen bzw. bei Personen mit einem hohen Risiko für eine FAP wird folgendes Vorgehen empfohlen:
- ab dem 10.Lebensjahr jährlich eine Rekto-Sigmoidoskopie (Spiegelung des Enddarmes)
- sobald erste Polypen auftreten jährlich eine komplette Koloskopie mit Entfernung der Polypen
- sobald die Polypen nicht mehr endoskopisch komplett entfernt werden können, wird eine Entfernung des Dickdarmes empfohlen, (Kolektomie, bei >=20 Polypen, Polyp >1 cm oder bereits vorliegende Dysplasie)
- Proktokolektomie, wenn Polypen auch im Rektum vorliegen
- alle 1-3 Jahre eine ÖGD (Ösophago-Gastro-Duodenoskopie)
- ab dem 25. Lebensjahr jährlich: Abdomensonografie, Endosonografie, ÖGD, Schilddrüsensonografie
Einen zum Thema Darmkrebs passenden Artikel aus dem Hamburger Ärzteblatt (Autoren: Prof. Dr. med. Sören Schröder, Frau Dr. med. Usha Peters, Dr. Dr. habil. Martin Keuchel) möchten wir Ihnen hier vorstellen:
[gview file=“http://www.dna-diagnostik.hamburg/wp-content/uploads/2015/04/kolorektale_polypen_s.pdf“]